scRNA seq with antibodies
Dr. Eric Chow, San Francisco, CA gives an overview of single cell sequencing, explains why this approach is useful, and talks through the leading methods.

Single cell sequencing, as the name implies, allows researchers to examine the genomic information for individual cells. This provides an opportunity to examine cell-to-cell differences and identify cell subtypes, which provides insight into how specific cells function within and respond to their environment.
This is an the best overview of single cell sequencing with a focus on RNA I found in 2025.

Lieven Gevaert, Bio-ir RUG 1996, Gentaur -Genprice Inc.
One can distinguish between plate-based, microfluidic-based, and combinatorial indexing methods.
Other approaches to single cell analysis don’t rely on RNA, including methods that use DNA, proteins, and antibodies.
- Benefits and limitations of analysis at the level of individual cells.
- Bulk vs. single cell analogy plate-based SMART-seq
- DropSeq
- Combinatorial Indexing
- Department of Biochemistry and Biophysics and the Director of the Center for Advanced Technology (CAT) at the University of California, San Francisco.
- The CAT provides resources for UCSF labs wishing to use next generation sequencing techniques and Chow’s research program strives to develop new applications for NGS in pathogen diagnostics.
- Chow received his BA in molecular biology from the University of California, Berkeley and his PhD in biochemistry from UCSF.
$ingle cell $equencing |
Single cell sequencing focusses on scRNA SEQ or RNA sequencing. |
| We'll cover plate-based methods, microfluidic methods, combinatorial indexing methods, and then talk about other single cell sequencing methods that don't rel. |
| necessarily relate to RNA. So, a big question that comes up is, why do single cell sequencing? |
| And a good analogy of this is this fruit
smoothie analogy. So, let's say that you're working with a blood sample, and
you want to take a look at the expression profile of. |
| Within that blood sample. So, blood is a complex mixture of many different cell types. You have B cells, T cells, macrophages, and neutrophils, |
| similar to a smoothie, where you have
lots of different types of fruit. And let's say you're really interested in
the characteristics of, say, a raspberry or an orange. |
| But instead of analyzing that raspberry or orange, you mix it up with a pineapple, a banana, and some strawberries, blend them all together, |
| and then that's what you're tasting.
It's gonna be very difficult to figure out what tastes are coming from that
raspberry. |
| And so, the same thing when you're trying to analyze data from a single cell versus a bulk sample. It's really advantageous if you can get data |
| specifically from individual cells, as opposed to making a bulk measurement. And one really cool thing about single cell sequencing |
| is that with the data you can identify lots of different cell types. And so, this is a representation of a. |
| of a data set from a blood sample. And in this case, many thousands of single cells were processed together, in parallel. |
| Each of those indic. individual single cell's transcripts were sequenced and analyzed. And you can detect up to, you know, |
| the 20,000 different genes that are present in the human genome. And so, it's really hard to depict data in 20,000 different dimensions, |
| so one common method is to collapse that down into two dimensions using dimensionality reduction. And there are many different methods, |
| which we won't be able to go over today, but you'll end up with a plot that looks like this, where each small dot represents a single cell, |
| and these dots are clustered, and cells that are clustered closer together are more similar to each other. |
| And so, you'll end up with these clusters of different cell groupings that are represented on this plot. |
| And so, this by itself isn't useful. You can tell that you have different cell types there, but you have no idea what they are. |
| But you can dig into the data. And. for instance, if you can go into each of these clusters and ask, what are the transcripts that differentiate, |
| say, this cluster at the top from this cluster next to it?, and you do this for the different genes, you can use this information to then classify |
| the different clusters, and identify them. So, for instance, in blue you have B cells; in red, you have activated CD8 T cells; |
| you have dendritic cells in this purple color; CD4 monocytes in green; NK cells in gray. |
| And, you know, the list goes on and on. You can identify lots of different cell types by looking at the transcriptional profiles |
within each of those clusters. And so,
if you've watched some of the flow cytometry videos on iBiology, you'll note
that you can get the same information |
| from flow cytometry, so, why do single cell sequencing? And one reason why is that these clusters can be further broken down |
| and analyzed by those genes. And so, again, we can make measurements on up to 20,000 differentially expressed genes |
| that are present. that might be present in samples, whereas with flow cytometry and mass cytometry, you might be limited to a few dozen markers, |
| and that's it. And so, for instance, if we were to break down this cluster, and ignore the rest of the clusters, |
| and just analyze this cluster and try to differentiate this a little bit more, and zoom in and re-cluster the data, |
| we can take that green cluster, re-cluster it to get some better separation, and then ask, what are the genes that separate these? And you can go through this process iteratively. |
| So, if you identify an interesting cluster, you can dig down to see whether a gene expression signature is specific for a subset within a cluster. |
| And this is really the power of single cell sequencing, is that you get a very high dimensional readout of the data that you can't get with other methods. |
| And so, the output of single cell sequencing methods has really increased significantly over the past decade. |
| So, initially, back in 2009 was the first study where they sequenced one cell, and that was it. |
| And over the years, you can see that there's been an exponential increase in the number of cells that have been processed in different studies, with each of these circles representing |
| a different publication and the number of cells analyzed. And so, we'll start looking at the techniques that were used, you know, about. uhh. you know, five years in. |
| ago and earlier, that really analyzed individual cells by sorting them into separate wells of a plate, |
| preparing libraries from each of those individual cells, and then sequencing them. The most common method was this plate-based SMART-seq technique. |
| And in this technique, you use a flow cytometer, and you sort your cells of interest, one cell into each well of a 96- or 384-well plate. |
| You lyse that cell, and then you do a reverse transcription reaction using a SMART-seq-style library preparation. And with this library preparation method, |
| you have an oligo-dT that'll prime a reverse transcription reaction off of mRNAs that are polyadenylated. |
| This oligo-dT has a handle on the end. And during the reverse transcription process, DNA gets made using RNA as a [template]. |
| And at the end, with certain reverse transcriptases, they'll add a couple of non-templated C bases. And these C bases are used in conjunction |
| with a template-switch oligo that has these G's at the end. So, this template-switch oligo, on top, |
| hybridizes onto these non-templated C's that got added, and the reverse transcriptase is able to use this template-switch oligo |
| as a template for further polymerization. And in the end, you wind up with a cDNA that has PCR handles on both ends. |
| These undergo amplification so that you get many, many copies of each cDNA, and then this goes into a sample preparation. |
| a DNA library preparation method to complete the library prep process. And that's depicted here. |
| And the most common one used is this Nextera DNA library preparation method, where you would have your amplified cDNA up here; the Nextera transposase will fragment that cDNA into smaller bits; |
| add partial Illumina adapters; and then, through PCR, you amplify up this material |
| and complete the library preparation process. And so, with those methods, you're able to process a few hundred, |
| maybe a few thousand samples with a lot of work, because you are processing each cell |
| in a separate tube of a well of a plate. And so, this required a lot of pipetting, a lot of manual work, |
| and it was fairly expensive, because you're essentially doing a separate sample. DNA library sample preparation for each cell. |
| And so, a few years ago, there were a few methods that came out that parallelized this process, taking advantage of different techniques |
| that we'll go into next. And the first one we'll cover is the method DropSeq. So, this is a method that uses microfluidics or essentially. |
| different methods of creating very, very small-volume droplets and doing molecular biology within those droplets. And in this method, you have a chip, |
| where you have beads that flow in from the left-hand side, cells will flow in from this first junction, and so they'll get mixed together. |
| In the next junction, you have oil that flows in. And this oil will cause this aqueous phase to pinch off and form an emulsion. |
| And because of the way the system works, you wind up with a lot of droplets at the end. Most of those droplets will be empty. |
| Some droplets will contain a bead; some droplets will contain a cell; and a small fraction will contain both a bead and a cell. |
| And that's depicted over here. And again, this is a small minority of the droplets that will contain both one cell and one bead. |
| What's special about these beads is that each of these beads has many barcoded oligo-dT oligos on the surface. |
| This will allow it to capture the RNA that comes from each of the cells that are co-encapsulated. And again, each of these beads |
| has a different barcode from all of the other beads. And so, what happens is you get capture of the RNA onto the beads. |
| After that, you pull down the beads out of the solution, and you do a reverse transcription reaction, |
| again with this template-switching process. And at this point, all the contents of each cell are stuck to one bead, |
| and all of those cDNAs have the same barcode because they're attached with the same bead and, again, that bead has many oligo-dT's |
| with that same barcode on there. You can amplify up the cDNA, just like we discussed before, |
| by using PCR. And then you can prepare a DNA sequencing library that'll sequence the ends of all of these transcripts. |
| And because we've tagged each of these transcripts with a barcode that corresponds to a cell, essentially we've been able to process |
| many, many cells in parallel in a couple of tubes. And with this method, you can sequence thousands, even tens of thousands, of samples, or cells, in a single sample, |
| using just a couple of tubes. So, you wouldn't need, you know, 30,000 different wells, |
| which would be incredibly laborious and logistically. just almost impossible to do. So, this was a commercial effort that came out of uhh. |
| that came out of an academic lab. And one issue with this method is that, again, most of those droplets contain nothing. |
| And only a small fraction of those droplets contains both one cell and one bead. And the reason for this is because |
| those cells and beads randomly get encapsulated into those droplets. And so, it looks something like this, |
| where you may have a lot of partitions that are empty, in gray, and then you have a fraction that have |
| a single bead or a single cell. And then, because of that random nature, although you don't have most of the wells or the droplets |
| filled with a cell or a bead, you do have some droplets that contain two or more beads, |
| or two or more cells. And so, you could do something where you increase the concentration of cells and beads |
| so that you get more single droplets that. droplets that contain a single cell or a single bead, but your doublet rate increases by a lot. |
| And there's no way to really distinguish this from the sequencing data. So, this is the reason why, with this DropSeq technique, |
| both the cells and beads are loaded at a low concentration -- to avoid those multiplets. |
| So, about a year or two after DropSeq came out, 10X Genomics, a commercial company, came up with a very, very similar method. |
| If you take a look at it, it looks extremely similar. You have barcoded beads that flow in from the side; cells and reagents come in; |
| and then those get mixed; and then oil comes in to generate an emulsion. But you can see, in this picture, |
| most of those droplets contain a bead, and that's because they are able to engineer both their microfluidic device and these beads |
| so that, you know, greater than 90% of the droplets contain one bead and only one bead. And so, this increased the efficiency by a lot, |
| because they don't have to load the beads at a low concentration. Another difference with this technique is that |
| inside of these droplets you don't just have capture, but you also undergo reverse transcription within these droplets, |
| which is one other difference. So, at the end of this reverse transcription reaction, you break the emulsion, |
| you take the cDNA and you amplify it up in a single tube, and then you construct your sequencing library, |
| again, in a single tube, and then you can sequence this on the sequencer. And depending on how many cells you load in the platform, |
| you can sequence up to 25,000 cells in each of the lanes of this chip. And this chip has 8 lanes on there, |
| so potentially, in a single chip, you could process 200,000 cells. And this process on the chip actually only takes 15 minutes. |
| The downstream portions over here take about half of a day, but that's pretty impressive that in essentially in one long day, |
| you could process 200,000 cells at a time -- or more if you ran multiple chips. So, another method to parallelize this process |
| of generating single cell sequencing libraries is to use microwells. So, instead of using an emulsion |
| to separate your cells from each other, you have a really, really tiny array of wells. So, each of these wells may only be |
| a few tens to a few hundred microns in diameter. And so, there are a couple of open source and also commercial platforms that do this, such as SeqWell, Celsee, |
| and one's from BD. And essentially, the way these work is that you have this microwell array, and there can be thousands, tens of thousands, |
| #NAME? |
| And these beads are engineered so that most of the time you get one and only one bead into a well. They kind of coat this array of microwells. |
| And then you apply your cells on top. In this case, in this example from Celsee, these cells are labeled in red so you can see them. |
| And again, in most of these wells, you have a blue colored bead that's present. And then the cells will just settle down into the wells. |
| And then you go through lysis and all the enzymatic reactions to tag the RNA from each of those cells |
| with a unique barcode, and go through this library preparation process to generate cells. And so, what's. to generate sample libraries. |
| And with some of these methods, they have wells, again, that have hundreds of thousands of wells, and potentially a million or more wells, on a chip |
| so that you can process many, many samples. So, within this past year, a paper came out that had |
| over a million cells processed in the study. And this [paper] actually used a method called combinatorial indexing, |
| which I'll go over next. And so, with combinatorial indexing, what happens is you do a lot of in situ reactions, |
| which means you use a cell as a compartment. And you're not working with individual cells at any given moment, |
| but you do multiple rounds of barcoding and mixing so that you generate these combinatorial barcodes |
| that will allow you to generate single cell data. And this is a method from an academic lab at the University of Washington |
| that used a combinatorial indexing method. And the way this works is that you have fixed cells, on the right -- so, these are fixed and permeabilized cells |
| so that reagents can flow in and out of the cells. You sort maybe a few dozen cells into each well of this 96-well plate. |
| So, you're not dealing with single cells. There are still multiple cells in each well of the plate. You undergo a reverse transcription reaction |
| with a barcoded oligo-dT. So, in each of these wells, you have a different barcoded oligo-dT. |
| So, all the cells within each well will have the same barcode at this step, okay? And this is depicted by these colors over here. |
| So, let's say you put. pulled from three wells, and so you have, you know, |
| many cells that have green, blue, or red. You combine these cells together or you shuffle them up, and you randomly redistribute them |
| into a new set of wells of a 96-well plate. At this step, you take your cDNA that's present in each of those cells, |
| and then you do a different reaction that will then add a second barcode. Again, each of the cells in each well |
| will receive the same barcode, but chances are each cell in this well came from a different well in this first plate. |
| And so, with this combination of barcodes -- you know, with 96 and 96, you have about 10 000 combinations -- |
| if you're only sequencing about a thousand cells, you don't have a very high chance of having two cells with the same barcode. |
| And this is essentially what the final library looks like. You have a RT barcode, or a reverse transcription barcode. You also have two different barcodes |
| that are in the Illumina adapters that can be used as well. So, you can potentially barcode up to three times |
| with this method. And the power of this technique really comes in as you add more of these levels. |
| So, for instance, again, if you had two sets of 96-well plates and you were barcoding them, |
| you have almost 10,000 combinations. If you were to work with 384-well plates, you'd have almost 150,000 combinations. |
| And it actually isn't ridiculous to work with several 384-well plates at each step. And so, if you had four 384-well plates at each step -- or 1536. combination of each step -- |
| you can generate about 2.4 million different combinations. And so, this is just with two rounds of barcoding. If you were to do three rounds of barcoding |
| and add another barcode -- in this case, 96 or 384 for a third level -- you really scale the numbers. |
| And so, in this case, I'm actually representing these numbers in millions now, just because the numbers would be too big to fit on this chart. |
| And so, in this case, if you were to do 96 x 96 x 96 with three levels, you'd have about a million combinations. |
| If you did 384 x 384 x 384, you'd have almost 60 million combinations. And so, this is the reason why this scales |
| really, really well, especially if you add that third level of barcoding. So, changing gears a little bit, |
| we focused mostly on RNA sequencing, but it is possible to do other things with single cell sequencing platforms. |
| And one of those is to quantify proteins. And so, you might ask, how do I quantify a protein with DNA sequencing? |
| And the method that's been used, called CITE-seq, is that you have your antibodies |
| that will bind onto proteins of interest. So, this is similar to an antibody that you might use for flow analysis, |
| but instead of conjugating a fluorophore to be read out by flow, or an isotope for CyTOF, |
| you conjugate on a DNA barcode. And so, the DNA barcode has a PCR handle at the end, a barcode that tells you which antibody it's attached to |
| -- again, this is just a DNA sequence barcode, so your A's, C's, G's, and T's; you just have some unique sequence for each antibody -- |
| and then a polyA tail. So, this polyA tail will allow this antibody sequence to get captured by the single cell sequencing methods, |
| and then you can read out the barcode, essentially as a. as a transcript that will tell you how many copies of this antibody |
| were bound, you know, to that particular cell. And so, this is just some data comparing RNA analysis with protein analysis, |
| just using human and mouse cells that have been kind of mixed together, to show you that the method works. |
| And there are currently panels of antibodies -- about 100 or even more antibodies -- that you can use in conjunction with single cell sequencing. |
| And the cool thing here is that you can get not just protein abundances for those proteins that you have antibodies for, |
| but you also get the transcriptional readout in terms of which genes are also turned on and off in each of these cells. |
| So, you get this kind of a. a multi-omic approach, where you can analyze both protein and RNA. So, one issue I told you about earlier |
| with some of these methods is this issue of multiplets. So, if we ignore just bead multiples |
| and just focus on cell multiplets, this is one reason why we're limited in terms of the number of cells that come out of these. |
| both these microfluidic and microwell platforms. And again, you don't want to load very many cells because you want most of these droplets |
| to be empty so that you avoid these doublets. So, again, if you were to increase the concentration of cells that you load in to maximize the number of singlets |
| -- so, for instance, over here you have four singlets -- you do increase the rate of your doublets -- so, these increase here and here. |
| Now, if there's some way to identify these doublets, you could remove them computationally from your data analysis. |
| And so, for instance, if you had two differentially labeled samples that you pooled together, |
| and then you flowed into your microfluidic device at a high concentration, you would get multiplets. |
| But if the multiplets came from two different cell types, and you can identify that, you can exclude those from analysis, |
| and then actually get more single cells for the same cost. And so, one. the first strategy to do this was a method called demuxlet. |
| And this is multiplexing by genotype. So, this group, here at UCSF, was working with different patient samples, |
| and with different humans, we all have different DNA sequences. And some of these sequences are found in our transcripts, |
| and those can be detected by single cell RNA sequencing. And so, in this example, they had eight different donors that had different genotypes, |
| depicted by different colors here, that got combined and then run in a single cell sequencing platform. |
| So, you get a whole bunch of single cell data. And if you take a look at each individual cell -- let's take a look at one cell, here -- |
| we see a certain genotype in the transcripts, and we can match that with one of the original donors. So, we can tell that this sample. |
| or, this cell came from this patient sample. And you can do this for most of the cells in. in the. in the data set. |
| What's really cool about this method is that if you have a doublet -- two cells in a single droplet -- |
| and you have eight different samples, chances are those two cells came from different samples, and they'll have two different genotypes. |
| So, if you analyze the data within those cells, if you see the presence of two different genotypes, you know that's a doublet, |
| and we can remove those from analysis. So, this allows you to bump up that concentration and get more single cell data |
| for the same cost. So, if you're not working with genetically distinct individuals, this method won't work, |
| and so there are a couple of non-genetic strategies that came out. The first one was from the same group that developed CITE-seq. |
| They realized that if they used an antibody that binds to some type of universal surface antigen on cells, they could take that antibody |
| and split it up into a couple of different aliquots. And with each aliquot, you can give it a different barcode. |
| Then, you can take your different samples and then add them to these different aliquots of antibodies to allow them to bind. |
| At that point, you can pool them all together, run them in the single cell sequencing platform, and you look for the presence |
| of which barcoded antibody showed up. And this will allow you to set. assign each of these individual cells |
| to a particular starting sample, because you knew which barcode. the antibody used for each sample. |
| And again, this also has the advantage where, if you have two or more cells in a droplet, you can detect those, |
| because you'll see two different barcode sequences assigned to a single droplet. And you can exclude that from analysis. |
| More recently, in collaboration with other group at UCSF, we developed a method that is a universal method |
that uses lipidated DNA that embeds into
the membranes of cells. So, this doesn't depend on the surface antigen.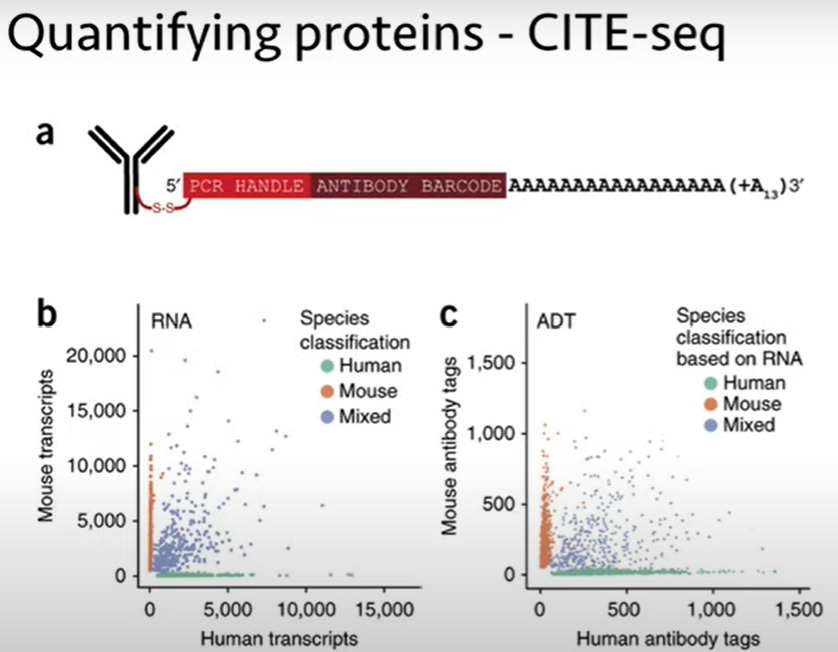 |
| It just depends on an accessible membrane that this barcoding DNA molecule can insert into. So, it inserts into the membrane of cells. |
| You can insert a different barcode into each pool of samples, and then pool them together. In this case, we were able to barcode |
| 96 different samples, pool them together, and we can identify all the singlets and the doublets. And you can see all the different 96 samples |
| represented here, in this plot. So, in conclusion, there are actually many different types of single cell sequencing. |
| I've only focused on RNA today, but there's DNA, there's protein, there's chromatin accessibility. |
| And many, many new methods coming out, you know, every few weeks. So, it's an exciting field to be in. |
| Single cell sequencing also generates high dimensional data, especially if you're looking at RNA, |
| because you're probing the quantity, or the expression level, of all 20,000 different genes that can be expressed in cells. And so, this can reveal biology |
| that's hidden from lower dimensional techniques. So, single cell sequencing does have some disadvantages. It is lower throughput and more expensive |
| compared to flow cytometry and mass cytometry. You know, with those methods, you can easily analyze hundreds of thousands |
| or millions of cells at a time. And I also didn't mention single cell analysis that doesn't use sequencing. |
| So, there are microscopy-based techniques that are out there as well that have some very interesting, you know, properties, |
| including the ability to give you some spatial information. So, if you're working with tissues, with most single cell sequencing techniques right now, |
| you have to dissociate those tissues into a single cell suspension and then run them. |
| So, you lose a lot of that spatial context that might be important for the biological question that you're trying to answer. |
| And some of these microscopy methods can offer you some additional information that might be valuable. And so, again, thank you for watching today. |

References
Cao, J. et al.. (2017). Comprehensive single-cell transcriptional profiling of a multicellular organism. Science, 357(6352), 661-667.
Kang, H.M. et al. (2018). Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nature biotechnology, 36(1), 89.
Macosko, E.Z. et al. (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell, 161(5), 1202-1214.
McGinnis, C.S., … & Chow, E.D. (2019). MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nature methods, 16(7), 619-626.
Stoeckius, M. et al. (2017). Large-scale simultaneous measurement of epitopes and transcriptomes in single cells. Nature methods, 14(9), 865.
Stoeckius, M. et al. (2018). Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome biology, 19(1), 1-12.
Svensson, V., Vento-Tormo, R., & Teichmann, S.A. (2018). Exponential scaling of single-cell RNA-seq in the past decade. Nature protocols, 13(4), 599-604.
Zheng, G.X. et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nature communications, 8(1), 1-12.